
Important to minimize degeneracy in a primer design
1. In the replication of ColE1 based plasmids, explain how copy number is normally regulated, and define and explain the consequences of inactivating mutations in:
a. ROM
b. RNA1
c. RNA2
d. PCNB
…Why might you want to temporarily reduce plasmid copy number in your cloning experiment?
2. Define and explain the basis for plasmid addiction of F plasmids. Speculate on 3 types (3 genes) of mutations that should eliminate this addiction.
3. Predict and explain the consequences (at least 2) of a mutation in RepE of the F plasmid that specifically blocked its ability to dimerize. What would the consequences be, of mutations in the incC locus?
4. You’ve ligated your favorite PCR fragment into a lacZ-based vector, along with controls, and transformed E coli to get the following Amp resistant colonies from equal aliquots of transformed cells, in expt A versus B versus C:
A B C
a. No DNA 0 0 0
b. Uncut vector tntc tntc tntc
c. Cut vector 3b 3b 180b
d. Cut vector + 0 ul PCR fragment 210b 5b 167b
e. Cut vector + 1 ul PCR fragment 177b, 43w 56w, 7b 193b, 25w
f. Cut vector + 3 ul PCR fragment 158b, 55w 219w,11b 188b, 63w
…where d,e,f had T4 DNA ligase added to the ligation mix; tntc = too numerous to count b = blue, w = white colonies Interpret your results, for experiments A, B, and C.
5. You have a stem cell from a patient with a known mutation in Gene X, which is the cause of the patients disease…5′-…CATCTGTGACGCTGTTTAGcTAGCAGTCAGCTA A, where “c” is the mutation, wild type is G. How could you use Crispr to correct that mutation in the stem cell, in a seamless manner, assuming you could introduce your construct into 20% of the cells in the population in vitro? What sequence would you use in your Crispr construct, what would you introduce into the stem cell, and how would you identify which transfectants had the correction?
6. You’re working in an organism in which the only tool is a multicopy shuttle plasmid with which you can transform your organism, and you’re looking for genes that are involved in regulating cell wall metabolism. Outline a function based strategy (a list of conceptual steps) you will take to identify these genes…other than searching by homology/whole genome sequencing. You are given a drug that blocks cell wall synthesis in your organism.
7. Before PCR technology was available, it was possible in yeast to snatch out specific genes from a genome, e.g. a mutant allele, using cloned wild type recombinant plasmids and homologous recombination. Using what you know from these, how could you most efficiently modify a yeast wild type cloned X to recover mutant gene X from yeast into E. coli?
8. You’ve transfected a neoR, tk+ vector into your mouse stem cell line. Explain how you will select for cells which have integrated the plasmid, and how you will further select against cells which have integrated by non-homologous recombination? Why is this dual selection needed? Describe and sketch outcomes of a PCR reaction you could run to convince yourself, A, that your homologous recombination was successful, and B, that no other non-homologous recombination events occurred?
9. Viral vectors are used for in vivo gene therapy because they offer efficient entry of DNA into target cells. Explain two problems with this approach. Explain how use of human stem cells may bypass the need for efficient transfection, and how you might use these for true disease gene correction.
10. In creating a synthetic genome, A: why use in vitro “stitching” methods or in vivo yeast recombination to merge synthetic sequences…why not just synthesize extremely long oligomers? B: for higher order “stitching” of very long sequences, would in vitro or in vivo (yeast) methods be better, and why? C: suppose you want to make a series of changes in a bacterial genome to test their effects on, say, virulence, but the species has no recombinant DNA tools and is difficult to manipulate. What approach could you take?
11. You perform peptide display pannings with your favorite ligand, only to find that the final peptides are as diverse as your initial input population. A. Why is this a problem. B. Think of and explain two different reasons why you might get this result. C. What could you do experimentally to distinguish which of your two explanations might the cause of the problem? Include examples of outcomes you’d get, depending on which explanation was the problem.
12. Cloning has a key advantage over PCR in that cloning from complex mixtures of many inserts allow separation of individual sequences, in the colonies or plaques. Propose a modification of PCR that would let you separate and recover large numbers of individual sequences amplified from an initially mixed sample, like a restriction digest or fragmented genome or a mixture of bacterial species.
13. Using a YEp library of wild-type S. cerevisiae genes, you did in vivo complementation in a mutant yeast host, and recovered 6 clones from transformants which had restored, normal phenotype. A. How could you be sure that the recovered clone was responsible for the normal, complemented phenotype? B. How will you identify the responsible genes on the 6 clones? C. When you disrupt the chromosomal gene corresponding to one of the 6 cloned genes in a wild type host, the resulting disruptant had a normal phenotype. Propose a mechanism for this unexpected result.
14. The yeast one-hybrid matchmaker system was used to identify some of the proteins in the yeast ORC complex. So, you decide to try this tool to identify mammalian ORC proteins. A. What will you use for your bait DNA sequence? B. Suppose you are unable to find any mammalian protein which, when fused to the activation domain, is able to activate the yeast reporter gene. Offer an explanation for this failure, other than inability of the hybrid proteins to express or to enter the nucleus….keeping in mind that the yeast ORC proteins were identified in this system.
15. Propose a strategy that uses colony filter hybridization, but which requires you to screen 30,000 colonies to have a reasonable chance of finding a few positive signals. Your strategy should use no more than 10 plates total, and at its conclusion would identify individual, pure colonies. Best answers give thoughtful consideration of the numbers.
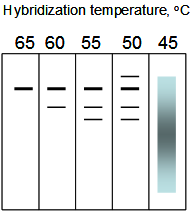
16. You have cloned gene X from your favorite organism, for which there is no genomic sequence available, and performed Southern blots of genomic DNA from the same organism, cut with EcoRI. You hybridize radiolabelled DNA from clone X to replicates of these blots, under the indicated hybridization temperatures, and see the following autoradiograms:
A. Interpret these observations. Best answers account for the data and offer explanations for what would generate the results.
B. Propose 2 ways of cloning these fragments, one using a cloned library and another exploiting the given gel observations. Sketch out the conceptual framework for your strategies.
17. Why do the AmpS ? AmpR and restriction site eraser methods for mutagenizing cloned genes require two sequential transformations into bacteria? Why are mutS strains used in the first transformations? What could you do experimentally to increase the chances that your clones will have the target mutation and not just the AmpR mutation? Why does EMILI need DpnI but GeneArt’s approach does not?
18. Why does genomic DNA cleavage via a designer ZCF or TALENS cutter, followed by non-homologous end joining, generate a site specific mutation (research question e.g. pubmed)? Suppose you want to generate a 57 kb deletion in a genome to test the hypothesis that the region is essential for virulence but not for growth…describe your approach to making this specific deletion.
19. In synthetic biology, graphically illustrate and explain how you control the relative order of genes you are stitching together, and the orientation of individual gene segments in that order. Illustrate and explain why cloning is usually necessary as an intermediate stage in constructing very large fragments.
20. Design a 21 base DNA primer that has the minimum amount of degeneracy, which will complement all possible encoding sequences anywhere along this protein sequence:
ASIDSAPLYSEMSPPRSDLIASSLTSPESIQMSVSGDVVGVNPYLNETETINFYDGYTSI
…why is it important to minimize degeneracy in a primer design?